留位費 (Deposit): HK$200/位
 【單基因遺傳病檢測(11+種) (HG-PMS-11+ )】
【單基因遺傳病檢測(11+種) (HG-PMS-11+ )】
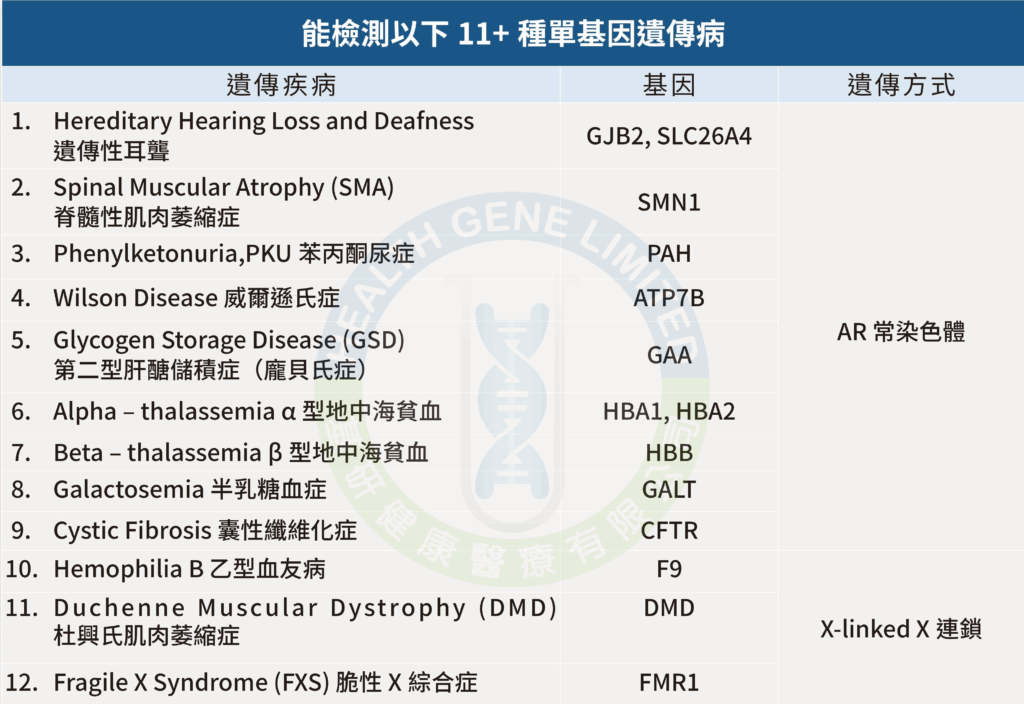
檢測12種遺傳病,14個基因
📍 價錢:HK$4,800/位
🛑 不用空腹
📄 報告時間:大約4-5星期
👩⚕️ 由醫生/專業醫護人員清楚解釋報告結果
🔄 檢查流程:登記 ➝ 醫護解釋檢查內容➝ 簽署同意書 ➝ 抽血 ➝全程大約30-45 分鐘
單基因遺傳病檢測的優點
- 全面 – 檢測全面,包括雙種突變類型
- 經濟 – 相比較於單個遺傳病的檢測,更加經濟
- 簡單 – 只需抽取5ml外周血
- 專業 – 全面的疾病數據庫,專業的基因檢測服務
- 準確度高達99%
除了一般有興趣人士,檢查亦適合︰
✔️適用於計劃生育的夫婦
✔️有家族病史的育齡夫婦
✔️準備進行人工輔助生殖的夫婦
✔️近親婚姻的夫婦
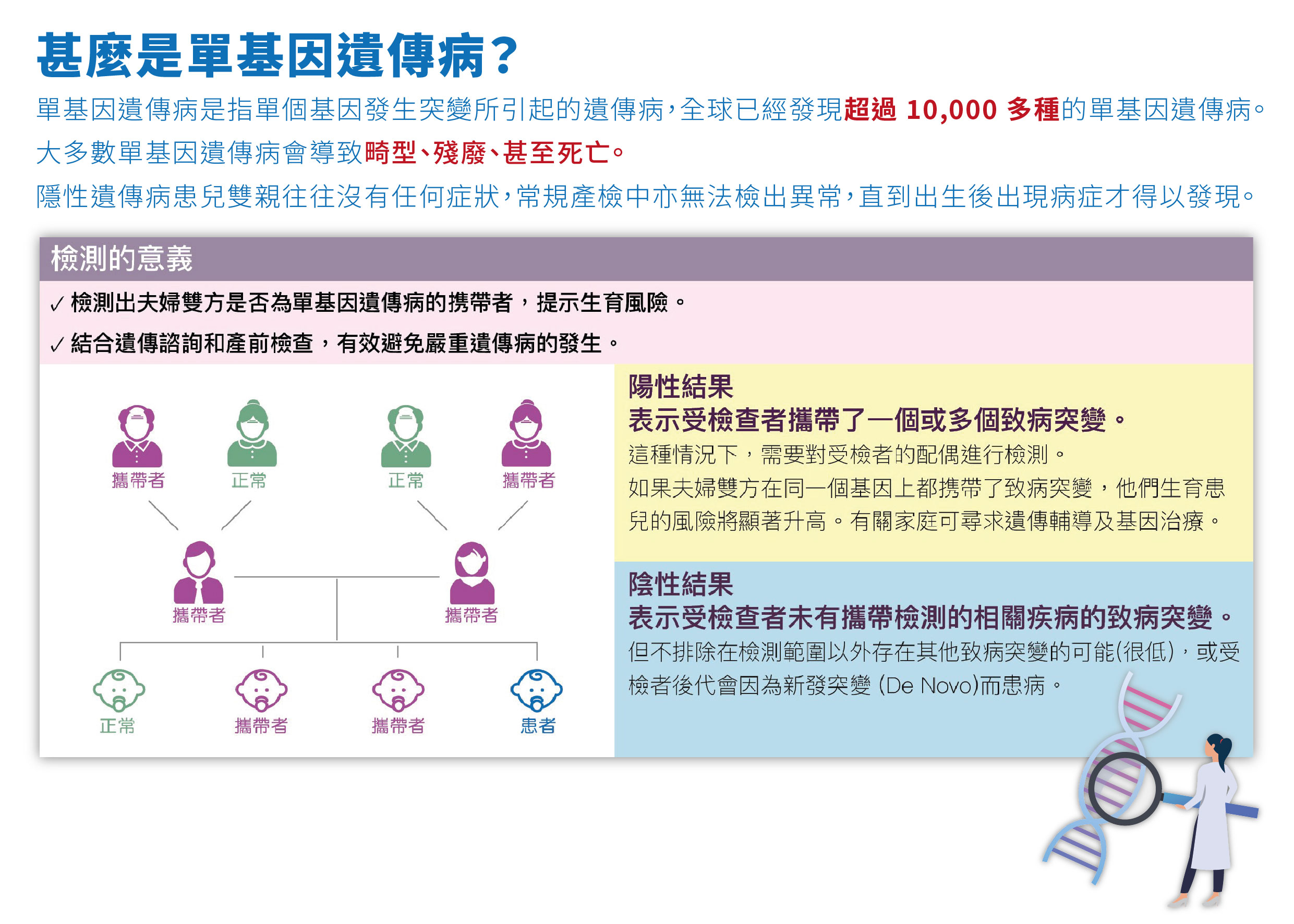
為何選擇單基因遺傳病檢測 ?
基於大多數人都不知道自己是遺傳性疾病的攜帶者,隱性遺傳病的人士往往沒有任何症狀, 常規產檢中亦無法檢出異常, 直到孩子出生後出現症狀才得以發現。因此「單基因遺傳病檢測11+ 種」能夠篩查12 種由5400 個以上的突變基因引起的遺傳性疾病, 相比於單一個遺傳病的檢測,「單基因遺傳病檢測11+ 種」更加經濟全面。
為何早期檢測是非常重要 ?
有家族病史的人士:
- 風險評估和管理:
對於有家族病史的人來說,進行單基因遺傳病檢測有助於早期發現基因疾病,及評估個人患某些遺傳疾病的風險,使其能夠根據需要進行生活方式調整或進行預防性篩查。
- 家庭計劃:
對於計劃擴大家庭的人來說,了解自身的遺傳風險可以影響他們的生育決策,並促使他們考慮其他選擇,例如代孕或是遺傳輔助技術。
想生育的夫婦:
- 遺傳風險輔助:
對於計劃懷孕的夫婦來說,早期檢測可以篩查任何可能性的遺傳疾病風險,使他們能夠做出知情的選擇。
- 生育決策支持:
檢測結果可以為夫婦提供關於懷孕的風險和選項的信息,使他們能夠共同決定最適合他們的生育計劃,從而減少未來可能的遺傳疾病風險。
遺傳疾病的簡介
- 遺傳性耳聾Hereditary Hearing Loss and Deafness
是指由基因突變引起的聽力障礙,可由父母遺傳給下一代,是新生兒最常見的先天性疾病之一。研究顯示,大約每 500 名新生兒中便有 1 名出現永久性聽力障礙,而當中相當部分與遺傳因素有關。
遺傳性耳聾主要由與聽覺系統相關的基因出現變異所致,可能影響內耳、聽覺神經或聲音傳導過程。這些基因突變可以在出生時已存在,也可能在成長過程中逐漸出現聽力下降。
在眾多與聽力障礙相關的基因中,常見致聾基因包括GJB2,SLC26A4。這些基因變異可導致不同程度的聽力損失,從輕度聽力下降至嚴重耳聾不等。
及早了解遺傳性聽力風險,有助於早期介入與適當醫療管理,對兒童語言發展及社交能力尤為重要。
- 脊髓性肌肉萎縮症 (Spinal Muscular Atrophy, SMA)
是一種由SMN1 基因的突變引起的遺傳性神經肌肉疾病。主要影響控制自主肌肉運動的運動神經元,導致肌肉無力和萎縮。 該病,該基因負責生成生存運動神經元蛋白(Survival Motor Neuron,SMN),該蛋白對於運動神經元的正常功能至關重要。
- 苯丙酮尿症 (Phenylketonuria, PKU)
是一種由PAH 基因的突變引起的遺傳性代謝疾病,患者體內缺乏苯丙氨酸羥化酶(PAH), 導致無法將食物中的苯丙氨酸(Phe)轉化為其他必需物質。苯丙氨酸的積累會對大腦造成損害,若未及時治療,可能引致智力和發育障礙。
未經治療的PKU 嬰兒在出生時可能無明顯異常,但在3 至6 個月大時可能開始對周圍環境失去興趣,並在1 歲時出現發育遲緩。其他症狀包括行為或社交問題、癲癇、小頭症、皮膚濕疹, 以及尿液、呼吸或皮膚散發出霉味。
- 威爾遜氏症 (Wilson Disease)
亦稱肝豆狀核變性(hepatolenticular degeneration),是一種由ATP7B 基因的突變引起罕見的常染色體隱性遺傳疾病,主要特徵是體內銅代謝異常,導致銅在肝臟、腦部和眼角膜等器官中異常積累。此病影響約每30,000 人中有1 人,男女發病率相等,通常在4 至40 歲之間發病。
患者可能出現以下症狀:
- 肝臟相關症狀:嘔吐、乏力、腹水、下肢水腫、黃疸和皮膚瘙癢等。
- 神經系統症狀:震顫、肌肉僵硬、言語困難、性格改變、焦慮,以及幻聽或幻視等。
- 眼部特徵:角膜邊緣出現Kayser-Fleischer 環,即銅在角膜邊緣沉積形成的金綠色或棕色環,是診斷該病的重要指標。
- 第二型肝醣儲積症(龐貝氏症)(Glycogen Storage Disease Type II, Pompe Disease)
又稱為龐貝氏症(Pompe Disease),是一種由於GAA (acid alpha-glucosidase) 突變引起的罕見常染色體隱性遺傳代謝病。基因突變導致體內缺乏或活性不足的酸性α- 葡萄糖苷酶。該酵素負責分解溶酶體內的肝醣,當酵素功能受損,肝醣便會在細胞內異常累積,尤其影響心臟與骨骼肌肉組織,進而造成多重器官損傷。
龐貝氏症依照發病年齡與嚴重程度可分為兩大類:
- 嬰兒型(Infantile-onset Pompe Disease):通常在出生後數月內出現症狀,包含嚴重的肌肉無力、呼吸困難、心肌肥厚與進行性心臟衰竭,若未及時治療,多在1 歲前死亡。
- 晚發型(Late-onset Pompe Disease):可於兒童或成人階段發病,症狀包括漸進性肌肉無力、呼吸肌無力(如橫膈膜)與活動能力下降,但心臟通常不受嚴重影響。
第二型肝醣儲積症(龐貝氏症)是一種可致命但可治療的罕見代謝疾病。透過新生兒篩查與基因檢測可實現早期診斷,配合酵素替代療法和持續追蹤,有助於改善患者預後,延長壽命並提升生活品質。
- α 型地中海貧血 (Alpha-thalassemia)
是一種遺傳性血液疾病,主要由HBA1 和HBA2 基因的突變或缺失引起,導致α- 珠蛋白鏈的合成減少或缺乏,進而影響血紅蛋白的正常功能。
α 型地中海貧血可分為以下類型:
- 靜默攜帶者(Silent Carrier):僅有一個α- 珠蛋白基因缺失或突變(-α/αα),通常無明顯臨床症狀,僅在血液檢查時可能發現輕度紅血球微小化。
- α地中海貧血特徵(α-thalassemia Trait):有兩個α- 珠蛋白基因缺失或突變,可分為順式(–/αα) 和反式(α/-α)。患者可能出現輕度貧血和紅血球微小化,但多數情況下無明顯症狀。
- 血紅蛋白H 病(Hemoglobin H Disease):三個α- 珠蛋白基因缺失或突變(–/-α),導致中度至重度貧血、脾臟腫大、輕度黃疸等症狀。部分患者可能需要輸血治療。
- 血紅蛋白Bart 氏胎兒水腫綜合症(Hb Bart’s Hydrops Fetalis Syndrome):四個α- 珠蛋白基因全部缺失或突變(–/–),為最嚴重的形式,導致胎兒期嚴重貧血、水腫,通常在出生前或出生後不久即死亡。
診斷主要依賴血液學檢查、血紅蛋白分析和分子遺傳學測試,以確定α- 珠蛋白基因的缺失或突變情況。對於輕度患者,可能無需特殊治療;而對於血紅蛋白H 病患者,可能需要定期監測、適當的輸血和脾臟切除等措施。建議高風險人群在孕前進行遺傳諮詢和基因篩查,以評估胎兒患病的風險,並提供相應的指導。
- β型地中海貧血 (Beta-thalassemia)
是一種遺傳性血液疾病,由HBB 基因的突變導致β- 珠蛋白鏈合成減少或缺失,進而影響血紅蛋白的功能。
分類:
- β地中海貧血主型(β-thalassemia major):患者通常在6 至24 個月大時出現嚴重貧血、蒼白、體重增加不良、發育遲緩、輕度黃疸和肝脾腫大等症狀。若未及時治療,可能導致生長遲滯、心臟擴張性心肌病、肝病和內分泌疾病等併發症。
- β地中海貧血中間型(β-thalassemia intermedia):患者的貧血程度較輕,可能無需定期輸血,但可能出現黃疸、膽石症、肝脾腫大、骨骼變形(如長骨畸形、特徵性顱面改變和骨質疏鬆)、腿部潰瘍、肺動脈高壓和血栓等併發症。
透過血液檢查可發現小細胞低色素性貧血,血紅蛋白分析顯示血紅蛋白A(HbA)減少或缺失, 血紅蛋白A2(HbA2)和胎兒血紅蛋白(HbF)增加。基因檢測可確定HBB 基因的突變,進一步確診。由於β 型地中海貧血為常染色體隱性遺傳病,建議高風險人群在孕前進行遺傳諮詢和基因篩查,以評估胎兒患病的風險,並提供相應的指導。
- 半乳糖血症 (Galactosemia)
是一種常染色體隱性遺傳性代謝疾病,最常見的類型為由 GALT 基因缺陷所引起的「經典型半乳糖血症」(Classic Galactosemia)。該基因位於 第9 號染色體長臂(9p13),負責編碼「半乳糖-1- 磷酸尿苷轉移酶」,此酵素為半乳糖代謝中不可或缺的關鍵。當酵素功能受損, 體內代謝路徑受阻,導致有毒代謝物累積並損害肝臟、腦部及其他器官。
嬰兒出生後攝取母乳或牛奶後,通常在數天內出現以下症狀:
- 餵食困難、嘔吐
- 肝功能異常(如黃疸、肝腫大)
- 體重不增、腹脹
- 凝血異常或出血傾向
- 嚴重者可能引發大腸桿菌敗血症,甚至危及生命
如未及時診斷並避免乳糖攝取,可能出現肝功能衰竭、腦損傷及永久性智能障礙。
乳糖血症是一種可透過新生兒篩查早期發現的嚴重代謝性疾病。雖然經適當飲食控制可避免急性併發症,但仍需長期追蹤與多學科照護以預防後續功能障礙。基因檢測在確診、家族帶因者篩查及婚前/孕前諮詢中均具有重要價值。
- 囊性纖維化症 (Cystic Fibrosis)
是一種複雜的遺傳性疾病,由CFTR 基因的突變引起,影響多個系統,主要影響呼吸系統和消化系統。患者體內的黏液變得濃稠且黏稠,導致肺部和消化道阻塞,增加感染風險。
主要症狀包括:
- 呼吸系統:持續性咳嗽(伴有黏液)、喘鳴、呼吸困難、頻繁的鼻竇和胸部感染。
- 消化系統:便秘、腹痛、腹脹、糞便油膩且難以沖洗。
其他可能的症狀包括兒童生長遲緩、成人體重無故下降、手指末端腫脹、汗液異常鹹等。
- 乙型血友病 (Hemophilia B)
亦稱為聖誕病(Christmas Disease),是一種由F9基因缺陷引起的遺傳性凝血功能障礙。該基因位於X 染色體上,負責編碼凝血因子IX。當F9 基因發生突變時,導致因子IX 的生成不足, 影響血液正常凝固功能。
此病主要以X 連鎖隱性遺傳方式傳播,故男性較易受影響,而女性則通常為無症狀的攜帶者。但部分女性攜帶者可能出現輕微出血傾向。
乙型血友病可分為以下三種嚴重程度:
- 重度( 因子IX 活性<1%): 患者可能自嬰兒期開始出現自發性出血,常見於關節(血腫) 和肌肉,若未及時治療,可能導致關節畸形和功能喪失。
- 中度( 因子IX 活性1%-5%): 通常在輕微創傷或手術後出現出血傾向。
- 輕度( 因子IX 活性5%-40%): 出血通常僅在重大手術或嚴重創傷後發生。
此外,患者可能出現鼻出血、牙齦出血、尿血或消化道出血等症狀。
- 杜興氏肌肉萎縮症 (Duchenne Muscular Dystrophy, DMD)
是一種嚴重的遺傳性神經肌肉疾病,由X 染色體上的DMD 基因突變引起,該基因負責編碼肌營養不良蛋白(dystrophin)。肌營養不良蛋白是肌肉細胞膜的重要結構蛋白,其缺乏導致肌肉纖維逐漸退化和壞死。主要影響男性,發病率約為每3,600 名男性新生兒中有1 例。
- 早期症狀(2 至3 歲): 患兒可能出現行走困難、經常跌倒、無法跑步或跳躍等運動發育遲緩的表現。
- 進展性肌肉無力: 隨著年齡增長,肌肉無力加重,通常在12 歲左右需依賴輪椅代步。
- 其他併發症: 心肌病、呼吸肌無力等,可能導致生命危險,患者平均壽命約為20 多歲。
- 脆性X 綜合症 (Fragile X Syndrome, FXS)
是一種由FMR1 基因變異引起的遺傳性疾病,該基因通常負責產生對大腦發育至關重要的FMRP 蛋白,是最常見的可遺傳性智力障礙之一。
症狀表現:
- 發育遲緩: 兒童可能在坐、走、說話等方面比同齡人晚。
- 學習困難: 在學習新技能時遇到挑戰。
- 社交和行為問題: 如避免眼神接觸、焦慮、注意力不集中、手部拍打等行為。
男性患者通常表現出中度至重度的智力障礙,而女性患者的症狀通常較輕。
預約流程

| AR常染色體疾病 | 1. Hereditary Hearing Loss and Deafness 遺傳性耳聾 |
|---|---|
| X-linked X連鎖疾病 | 10. Hemophilia B 乙型血友病 |