
Reservation Deposit: HK$200
Checkup Location: Suite 1424-1425, 14/F, Ocean Centre, 5 Canton Road, Tsim Sha Tsui, Kowloon
helthgene
Skip to content$4,800.00
Reservation Deposit: HK$200
Checkup Location: Suite 1424-1425, 14/F, Ocean Centre, 5 Canton Road, Tsim Sha Tsui, Kowloon
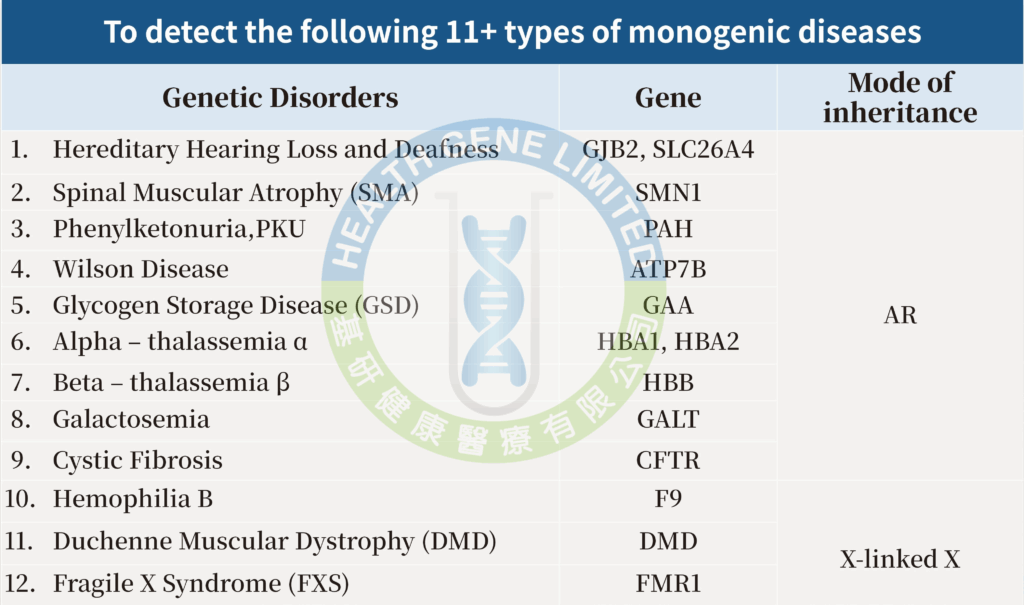
【Monogenic Screening (11+ Types) (HG-PMS-11+ )】
 Detect 12 types of genetic diseases, 14 genes.
Detect 12 types of genetic diseases, 14 genes.
📌 Price: HK$4,800 per person
🛑 No fasting required
🕐 Report turnaround: Approximately 4-5 weeks
👩⚕️ Report Explanation: Explained by GP or medical staff
🔄 Process: Registration ➝ Medical staff explain the details of this test➝ sign consent form ➝ Blood draw➝ around 30-45 mins
✔️ Couples planning for family planning
✔️ Reproductive-age couples with a family history of disease
✔️ Couples preparing for assisted reproductive technologies
✔️ Couples in consanguineous marriages
According to the World Health Organisation (WHO):
Since most people are unaware of being carriers of genetic diseases, individuals with recessive genetic diseases often show no symptoms. Routine prenatal tests also cannot detect abnormalities, leading to the discovery of such conditions only when symptoms appear in the child after birth. Therefore, the “Monogenic Screening (11+ Types)” can screen for 12 hereditary diseases caused by over 5,400 mutation variants. Compared to testing for a single genetic disease, the ” Monogenic Screening Test (11+ Types)” is more cost-effective and comprehensive.
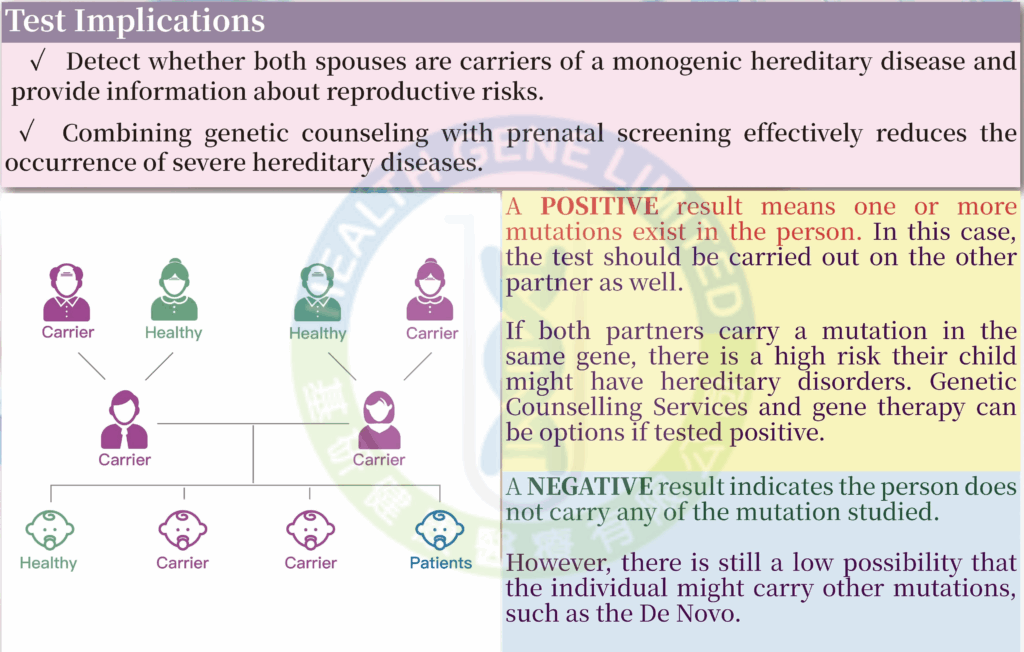
For individuals with a family history of genetic diseases:
For couples planning to have a baby:
Hereditary hearing loss and deafness refer to hearing impairments caused by genetic mutations that can be passed from parents to their offspring. It is one of the most prevalent congenital conditions in newborns. Research indicates that approximately 1 in every 500 neonates experiences permanent hearing loss, a significant portion of which is attributed to genetic factors.
This condition primarily results from mutations in genes associated with the auditory system, which may affect the inner ear, the auditory nerve, or the process of sound conduction. These genetic variants may be present at birth (congenital) or manifest as progressive hearing transition during childhood or later in life.
Among the numerous genes linked to hearing impairment, GJB2 and SLC26A4 are identified as common causative genes. Mutations in these genes can lead to varying degrees of hearing loss, ranging from mild impairment to profound deafness.
Early identification of genetic risks for hearing loss is crucial for timely intervention and appropriate medical management. This is particularly vital for a child’s linguistic development and social integration.
Spinal Muscular Atrophy (SMA) is a hereditary neuromuscular disorder caused by mutations in the SMN1 gene. This condition primarily affects motor neurons that control voluntary muscle movement, leading to muscle weakness and atrophy. The SMN1 gene encodes the Survival Motor Neuron (SMN) protein, which is essential for the normal function and survival of motor neurons.
Phenylketonuria (PKU) is a genetic metabolic disorder caused by mutations in the PAH gene, resulting in a deficiency of the enzyme phenylalanine hydroxylase (PAH). This enzyme is necessary for converting the amino acid phenylalanine (Phe), found in food, into other essential substances. Without proper enzyme function, phenylalanine accumulates in the body, potentially causing brain damage. If left untreated, PKU can lead to intellectual disability and developmental delays.
Infants with untreated PKU may appear normal at birth, but signs of developmental concern often begin between 3 to 6 months of age, such as reduced interest in surroundings, with developmental delays becoming more noticeable by around one year of age. Additional symptoms can include behavioral or social issues, seizures, microcephaly, eczema, and a musty odor in the urine, breath, or skin.
Also known as hepatolenticular degeneration, Wilson Disease is a rare autosomal recessive genetic disorder caused by mutations in the ATP7B gene. This condition leads to impaired copper metabolism, resulting in abnormal accumulation of copper in organs such as the liver, brain, and corneas. It affects approximately 1 in 30,000 individuals, with equal incidence in males and females, and typically presents between the ages of 4 and 40.
Patients may exhibit a range of symptoms, including:
Also known as Pompe Disease, Glycogen Storage Disease Type II is a rare autosomal recessive metabolic disorder caused by mutations in the GAA gene, which encodes the enzyme acid alpha-glucosidase. This enzyme is essential for breaking down glycogen within lysosomes. When the enzyme is deficient or non-functional, glycogen accumulates abnormally inside cells, particularly affecting cardiac and skeletal muscles, ultimately leading to multi-organ damage.
Pompe Disease is classified into two major forms based on the age of onset and disease severity:
Symptoms typically appear within the first few months of life and may include severe muscle weakness, respiratory difficulties, hypertrophic cardiomyopathy, and progressive heart failure. Without timely treatment, affected infants often die before the age of one.
This form may present during childhood or adulthood. It is characterized by progressive muscle weakness, including weakness of respiratory muscles such as the diaphragm, and a gradual decline in mobility. Unlike the infantile form, cardiac involvement is generally not prominent.
Pompe Disease is a potentially fatal but treatable condition. Early diagnosis through newborn screening and genetic testing enables timely intervention. Enzyme replacement therapy (ERT), combined with regular monitoring, can significantly improve prognosis, extend lifespan, and enhance quality of life for affected individuals.
Alpha-thalassemia is an inherited blood disorder primarily caused by mutations or deletions in the HBA1 and HBA2 genes, leading to reduced or absent production of alpha-globin chains. This impairs the normal function of hemoglobin and affects red blood cell development.
Alpha-thalassemia can be categorized into several clinical forms based on the number of affected alpha-globin genes:
Diagnosis relies on hematological tests, hemoglobin electrophoresis or HPLC, and molecular genetic analysis to detect alpha-globin gene deletions or mutations. Mild cases may not require specific treatment, while patients with Hemoglobin H Disease may need regular monitoring, blood transfusions, and possibly splenectomy.
Genetic counseling and carrier screening are recommended for individuals at high risk, particularly before pregnancy, to assess the potential risk of having an affected child and to provide informed reproductive guidance.
Beta-thalassemia is an inherited blood disorder caused by mutations in the HBB gene. These mutations lead to a reduction or total absence of β-globin chain synthesis, which subsequently impairs the function of hemoglobin.
The severity of the condition is generally categorized into two primary clinical forms:
Diagnostic evaluation typically involves a Complete Blood Count (CBC), which reveals microcytic hypochromic anemia. Hemoglobin analysis further identifies a reduction or absence of Hemoglobin A (HbA), alongside an increase in Hemoglobin A2 (HbA2) and Fetal Hemoglobin (HbF). Definitive diagnosis is confirmed via genetic testing to identify specific mutations in the HBB gene.
As beta-thalassemia follows an autosomal recessive inheritance pattern, it is highly recommended that individuals in high-risk populations undergo pre-pregnancy genetic counseling and screening. This helps assess the risk of transmission and provides prospective parents with necessary medical guidance.
Galactosemia is an autosomal recessive metabolic disorder, the most common form being Classic Galactosemia, caused by defects in the GALT gene. This gene is located on the long arm of chromosome 9 (9p13) and encodes the enzyme galactose-1-phosphate uridylyltransferase, a critical component in the metabolic pathway of galactose. When this enzyme’s function is impaired, the metabolic pathway is obstructed, leading to the accumulation of toxic metabolites that damage the liver, brain, and other vital organs.
Symptoms typically manifest within a few days after an infant begins consuming breast milk or dairy-based formula:
Note: Failure to diagnose the condition promptly or avoid lactose intake can result in hepatic failure, brain damage, and permanent intellectual disabilities.
Galactosemia is a serious metabolic disease that can be detected early through newborn screening. While strict dietary control (a lactose-free diet) can prevent acute complications, patients require long-term monitoring and multidisciplinary care to mitigate subsequent functional impairments, such as speech delays or ovarian insufficiency.
Genetic testing is of significant clinical value for:
Cystic Fibrosis is a complex genetic disorder caused by mutations in the CFTR gene. It is a multisystemic condition that primarily affects the respiratory and digestive systems. In affected individuals, the body produces abnormally thick, viscous mucus that leads to obstructions in the lungs and digestive tract, significantly increasing the risk of chronic infections.
Typically involves the following:
Other Clinical Indicators:
Hemophilia B, also known as Christmas Disease, is a hereditary coagulation disorder caused by a deficiency or defect in the F9 gene. Located on the X chromosome, this gene encodes Coagulation Factor IX (FIX). Mutations in the F9 gene result in insufficient production of functional Factor IX, thereby impairing the blood’s ability to clot effectively.
The condition primarily follows an X-linked recessive inheritance pattern. Consequently, males are predominantly affected, while females typically serve as asymptomatic carriers. However, some female carriers may exhibit a mild bleeding tendency if their Factor IX levels are lower than normal.
Hemophilia B is classified into three levels of clinical severity based on the residual activity of Factor IX:
In addition to musculoskeletal bleeding, patients may present with:
Duchenne Muscular Dystrophy (DMD) is a severe, hereditary neuromuscular disorder caused by mutations in the DMD gene located on the X chromosome. This gene is responsible for encoding dystrophin, a critical structural protein that maintains the integrity of the muscle cell membrane (sarcolemma). A deficiency in dystrophin leads to progressive degeneration and necrosis of muscle fibers. DMD primarily affects males, with an incidence of approximately 1 in 3,600 male live births.
The disease is characterized by progressive muscle wasting, typically following a predictable chronological course:
Fragile X Syndrome (FXS) is a genetic disorder caused by mutations in the FMR1 gene. This gene is responsible for producing the FMRP protein, which is critical for normal brain development. FXS is one of the most common forms of inherited intellectual disability.
Symptoms:
Key Observations:
Booking Procedure
 https://www.healthgene.com.hk/wp-content/uploads/2019/07/flow-en.png" width="1001" height="381" alt="" title="flow" />
https://www.healthgene.com.hk/wp-content/uploads/2019/07/flow-en.png" width="1001" height="381" alt="" title="flow" />For more details on the program, please click check-up items.
[/fusion_imageframe]
| AR常染色體疾病 | 1. Hereditary Hearing Loss and Deafness遺傳性耳聾 |
|---|---|
| X-linked X連鎖疾病 | 10. Hemophilia B 乙型血友病 |






